
Stanowisko Zespołu Ekspertów ds. Bioetycznych KEP w sprawie wentylacji płuc w SMA
Zespół Ekspertów ds. Bioetycznych KEP przyjrzał się aspektom etycznym ogólnych zasad postępowania z chorym na rdzeniowy zanik mięśni typu 1.

zdjęcie: www.episkopat.pl
2019-03-19
Żadna rodzina, w której jest chore dziecko na SMA, nie może czuć się osamotniona i pozbawiona pomocy – czytamy w wydanym 19 marca br. Stanowisku Zespołu Ekspertów ds. Bioetycznych KEP w sprawie wentylacji płuc w rdzeniowym zaniku mięśni typu 1. Stanowisko w imieniu zespołu podpisał jego przewodniczący – bp dr hab. Józef Wróbel, prof. KUL.
W pierwszej części stanowiska opisana została aktualna wiedza medyczna na temat rdzeniowego zaniku mięśni (SMA – ang. spinal muscular atrophy). „Przyjmuje się, że w Europie jest to liczba 12-18 mln. Ogólna chorobowość występowania waha się w przedziale 1,31-1,87 na 100 tys. osób. (…) W Polsce SMA ujawnia się w 1 przypadku na 5000-7000 urodzeń. Mając na uwadze wszystkie typy SMA, liczba chorych w Polsce wynosi mniej niż 1000” – czytamy w dokumencie.
Zespół Ekspertów ds. Bioetycznych KEP porusza aspekty etyczne ogólnych zasad postępowania z chorym na SMA. „Żadna rodzina, w której jest chore dziecko na SMA, nie może czuć się osamotniona i pozbawiona pomocy” – podkreślają eksperci.
W Stanowisku zaznaczono, że „obowiązkiem spoczywającym na rodzicach (prawnych opiekunach) i opiekunach medycznych jest ochrona życia i umacnianie go, jednak bez stosowania terapii daremnej”.
Autorzy dokumentu podkreślają, że „Standardy opieki nad chorym cierpiącym na SMA są wciąż udoskonalane. Z każdym rokiem jest ona efektywniejsza i daje większe szanse na przedłużenie życia, a być może i zupełne wyleczenie, ponieważ w ostatnich latach dokonał się przełom związany ze wspomnianym wyżej lekiem (Nusinersenem), który zmniejsza ryzyko śmierci i opóźnia wprowadzenie wentylacji przewlekłej. Badania ostrożnie sugerują, że istnieje duże prawdopodobieństwo wyleczenia choroby w przypadku włączenia leku w okresie przedobjawowym. Nie wiadomo jeszcze, jaka będzie skuteczność leczenia w przypadku dziecka, które miało już objawy w chwili rozpoczęcia terapii”.
Dokument jest odpowiedzią na pytania etyczne dotyczące decyzji podjęcia sztucznej wentylacji u dziecka chorego na rdzeniowy zanik mięśni typu 1. Zespół Ekspertów ds. Bioetycznych KEP przedstawił uszczegółowioną aplikację zasad ogólnych w terapii dzieci chorujących na tę chorobę. Zasady te mogą stanowić model postępowania w przypadku podobnych chorób.
Pełna treść dokumentu:
Stanowisko Zespołu Ekspertów ds. Bioetycznych KEP w sprawie wentylacji płuc w rdzeniowym zaniku mięśni typu 1
1. W dniu 27 czerwca 2018 r. Zespół Ekspertów Konferencji Episkopatu Polski ds. Bioetycznych, odpowiadając na prośby płynące z różnych środowisk i grup społecznych, wydał dokument O terapii daremnej (uporczywej) chorych poddawanych intensywnej terapii. Odwołując się do naczelnej zasady bioetyki personalistycznej, jaką jest konieczność poszanowania życia i godności osobowej człowieka, wskazano, że terapia daremna wyklucza przyśpieszenie śmierci poprzez działania eutanatyczne, jak i jej sztuczne opóźnianie przez możliwe działania medyczne.
2. Przypomniane w tym dokumencie zasady znajdują zastosowanie w podejmowaniu decyzji dotyczących sposobu leczenia pacjentów chorujących na różne rodzaje chorób. W odpowiedzi na pytania etyczne dotyczące decyzji podjęcia sztucznej wentylacji u dziecka chorego na rdzeniowy zanik mięśni typu 1. Zespół Ekspertów przedstawia uszczegółowioną aplikację zasad ogólnych w terapii dzieci chorujących na tę chorobę. Poniższe zasady mogą stanowić model postępowania w przypadku podobnych chorób.
Aktualna wiedza medyczna na temat Choroby pacjenta opisanego w liście oraz możliwości jej leczenia
3. Rdzeniowy zanik mięśni (SMA – ang. Spinal Muscular Atrophy) to rzadka genetyczna choroba, nerwowo-mięśniowa, ale najczęstsza z rzadkich, dziedziczona w sposób autosomalny recesywny. Około 1 na 50 osób jest nosicielem tej choroby. Przyjmuje się, że w Europie jest to liczba 12-18 mln. Ogólna chorobowość występowania waha się w przedziale 1,31-1,87 na 100 tys. osób. Każdego roku z tą chorobą rodzi się 8,5-10,3 dzieci na 100 tys. żywych urodzeń. Oboje rodzice dzieci chorych na SMA są przeważnie bezobjawowymi nosicielami choroby. Gdy ojciec i matka są nosicielami, w przypadku każdej ciąży zachodzi prawdopodobieństwo wystąpienia choroby w 25%, bezobjawowego nosicielstwa w 50% i braku choroby w 25%. W Polsce SMA ujawnia się w 1 przypadku na 5000-7000 urodzeń. Mając na uwadze wszystkie typy SMA, liczba chorych w Polsce wynosi mniej niż 1000. Dzięki wynikom badań dr Judith Melki z Francji w 1995 r. stwierdzono, że chorobę powodują mutacje genów SMN1 i SMN2 zlokalizowanych w chromosomie 5. odpowiedzialnych za kodowanie białka SMN (ang. survival motor neuron), niezbędnego do funkcjonowania neuronów ruchowych. Jego niedobór sprawia, że mięśnie ciała słabną i stopniowo ulegają zanikowi. SMA jest jedną z najczęstszych genetycznie uwarunkowanych przyczyn śmierci niemowląt i małych dzieci. W praktyce stosowany jest umowny podział SMA na cztery typy oraz dodatkowo typ 0 (nie uwzględniony w tabeli). Klasyfikacja ta opiera się na neurologicznej ocenie zaawansowania choroby u pacjenta.
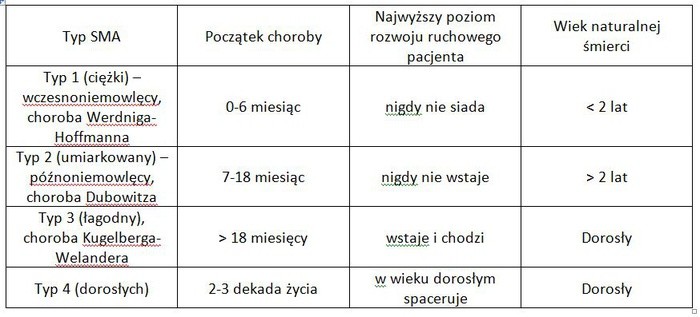
4. Dzieci chorych na SMA typu 1. sprawiają najwięcej dylematów anestezjologom-pediatrom i innym specjalistom pediatrii współpracującym z nimi. SMA typu 1. (nigdy niesiedzący) nazywana była dawniej „chorobą Werdniga-Hoffmanna”. U dzieci z podtypem 1a zgon następuje przed ukończeniem 2. roku życia, natomiast z 1b – po ukończeniu 2. roku życia. Jest to postać najcięższa i o najgorszym rokowaniu. U tych pacjentów objawy osłabienia mięśni pojawiają się nagle przed szóstym miesiącem życia, a często znacznie wcześniej. Stan dziecka pogarsza się bardzo szybko, stwierdza się u niego uogólnioną wiotkość, osłabienie siły mięśni, fascykulacje języka, trudności ze ssaniem i połykaniem, arefleksje lub hiporefleksje, osłabienie krzyku i oddechu. Dziecko nie jest w stanie unieść główki i nigdy nie będzie siedzieć bez podparcia. Szybkiemu osłabieniu ulegają mięśnie kończyn, tułowia, płuc i przełyku, co zazwyczaj prowadzi do niewydolności oddechowej i utraty zdolności przełykania. Ryzyko śmierci jest bardzo wysokie, a przewidywana długość życia wynosi mniej niż dwa lata. Dlatego utrzymanie dziecka przy życiu wymaga często specjalistycznej opieki na najwyższym poziomie, co jest trudne nawet w szpitalu III stopnia referencyjności. Natomiast choroba przebiega łagodniej u pacjentów z SMA typu 3. (długość życia pozostaje niezmieniona lub nieznacznie skrócona) lub typu 4 – długość życia pozostaje niezmieniona. Osłabienie mięśni następuje powoli, a chory stopniowo traci możliwość samodzielnego poruszania się.
5. Dotychczas w neurologii dziecięcej SMA typu 1. uważano za chorobę nieuleczalną, a po wykonaniu niezbędnej diagnostyki dziecko przekazywano pod opiekę hospicyjną. W ostatnich latach dokonał się jednak przełom związany z wykorzystaniem w leczeniu leku Nusinersen (nazwa handlowa – Spinraza). Badania kliniczne potwierdziły jego skuteczność polegającą na zwiększeniu ilość białka SMN produkowanego przez gen SMN2, identycznego oraz pełnowartościowego, jak w przypadku białka produkowanego przez gen SMN1, które przywraca funkcjonowanie neuronów i (lub) zapobiega ich obumieraniu. Nusinersen dopuszczony został do leczenia SMA w USA (24.12. 2016) oraz w UE (30.05.2017). Nusinersen podawany jest dooponowo przez nakłucie lędźwiowe. U dzieci, które urodziły się z poważnym niedoborem białka SMN, może jednak nie wystąpić znacząca poprawa stanu zdrowia. Rozpoczęte leczenie powinno być kontynuowane do końca życia. Według zaleceń firmy BIOGEN, na początku terapii podawana jest dawka wysycająca w dniach: 0., 14., 28. oraz 63. Dawkę podtrzymującą należy podawać raz na 4 miesiące.
6. Terapia ta jest jednak bardzo kosztowna i wynosi ok. 90 tys. euro za dawkę, natomiast koszt rocznej terapii to ok. 540 tys. euro w pierwszym roku i 270 tys. euro w kolejnych latach. Koszt tego leczenia i powiązanych z nim innych terapii jest poza możliwościami finansowymi zwykłej rodziny. Stąd też obecnie cała nadzieja jest w pomocy darczyńców oraz organizacji rodzin dzieci chorych na SMA. Należy ufać, że wkrótce rodziny chorych otrzymają również pomoc ze strony państwa, co zapewniłoby równy dostęp do tego leczenia.
7. Wiele dzieci było jednak już poddanych terapii Nusinersenem jeszcze przed rejestracją leku. Odbyło się to w ramach Programu Dostępu Rozszerzonego – EAP (expanded access program) dotyczącego chorób rzadkich. W Europie do 2018 roku przystąpiło do niego 600 osób; były to niemal wyłącznie dzieci. Według danych z lutego 2017 roku w Polsce włączono do niego dziesięcioro dzieci z SMA typu 1. Obecnie lek podawany jest 30 chorym w trzech ośrodkach: w Instytucie „Pomnik Centrum Zdrowia Dziecka” w Warszawie – 17 dzieci, Górnośląskim Centrum Zdrowia Dziecka w Katowicach – 6 dzieci oraz Uniwersyteckim Centrum Klinicznym w Gdańsku – 7 dzieci.
8. W leczeniu podejmowane były również próby z innymi środkami farmakologicznymi i biotechnologiami, takimi jak:
– Risdiplam (RG7916, RO7034067) i BRANAPLAM (LMI070, NVS-SM1), to lekarstwa eksperymentalne, których zadaniem jest produkcja normalnej ilości białka SMN.
– AVXS-101 jest obecnie jedyną udoskonalaną terapią genową w SMA. Działa poprzez wprowadzenie do komórek sekwencji odpowiadającej genowi SMN1 (tzw. transgenu) z pomocą nośnika, którym jest kapsyd wirusa AAV9.
– Reldesemtiv (znany również pod nazwą: CK-2127107, CK-107) jest szybkim aktywatorem troponiny – białka regulującego pracę mięśni poprzecznie prążkowanych i mięśnia sercowego. Lek poprawia funkcję mięśni, ale nie leczy wady genetycznej powodującej SMA.
– Terapia komórkami macierzystymi; w niektórych ośrodkach badań zaniechano, ponieważ uznano, że podawanie tych komórek jest nieskuteczne.
9. W opiece medycznej zalecane są międzynarodowe wytyczne Consensus Statement for Standard of Care in Spinal Muscular Atrophy (2005-2007), opracowane przez ekspertów pod kierunkiem prof. Thomasa Sejersena. Uaktualnione standardy wydane zostały w 2018 r. pod tytułem Diagnosis and Management of Spinal Muscular Atrophy. Dokument ten w części pierwszej omawia diagnostykę, rehabilitację, opiekę ortopedyczną i żywienie, natomiast w części drugiej prezentuje opiekę oddechową, intensywną terapię, leczenie, suplementację, szczepienia oraz etykę. Według tych założeń opieka nad chorym z SMA powinna być koordynowana przez neurologa i mieć charakter wielodyscyplinarny, łącząc pracę specjalistów z zakresu: pulmonologii (w Polsce głównie anestezjologii), ortopedii, dietetyki, fizjoterapii, pielęgniarstwa, terapii zajęciowej, psychologii, pedagogiki, logopedii oraz dostępnych technologii wspomagających funkcje życiowe pacjenta.
Ogólne zasady postępowania z chorym na SMA – aspekty etyczne
10. Żadna rodzina, w której jest chore dziecko na SMA, nie może czuć się osamotniona i pozbawiona pomocy.
11. Obowiązkiem spoczywającym na rodzicach (prawnych opiekunach) i opiekunach medycznych jest ochrona życia i umacnianie go, jednak bez stosowania terapii daremnej.
12. O zwyczajności bądź nadzwyczajności środków terapeutycznych może współdecydować także ich koszt oraz możliwości ekonomiczne poszczególnych osób lub całego społeczeństwa. W tym kontekście należy zauważyć, że niepodjęcie leczenia, którego koszt znacznie przewyższa te możliwości, nie powoduje zaciągnięcia winy moralnej. Z drugiej strony, zjawiskiem prawdziwie pięknym i szlachetnym są postawy solidarności społecznej z chorymi i ich rodzinami wyrażające się w gromadzeniu środków koniecznych do podjęcia i kontynuacji terapii.
13. Standardy opieki nad chorym cierpiącym na SMA są wciąż udoskonalane. Z każdym rokiem jest ona efektywniejsza i daje większe szanse na przedłużenie życia, a być może i zupełne wyleczenie, ponieważ w ostatnich latach dokonał się przełom związany ze wspomnianym wyżej lekiem (Nusinersenem), który zmniejsza ryzyko śmierci i opóźnia wprowadzenie wentylacji przewlekłej. Badania ostrożnie sugerują, że istnieje duże prawdopodobieństwo wyleczenia choroby w przypadku włączenia leku w okresie przedobjawowym. Nie wiadomo jeszcze, jaka będzie skuteczność leczenia w przypadku dziecka, które miało już objawy w chwili rozpoczęcia terapii.
Wyjaśnienia szczegółowe
14. Opis przypadku: Rodzice zostali poinformowani wcześniej (z zagranicznego ośrodka) przez lekarzy, że takiego dziecka się nie intubuje, bo to tylko przedłuża jego cierpienie, a jakość życia jest żadna. Ponieważ stan chłopca się pogorszył, przesłali go do Polski, gdzie został przyjęty na oddział intensywnej terapii (3. stopień referencyjności). Rodzice nie zgodzili się na żadne formy inwazyjne – intubację, wentylację, a poinformowani o niewydolności oddechu oraz w następstwie krążenia, która może nastąpić w każdej chwili, napisali oświadczenie, że nie zgadzają się na resuscytację. Jak należy postąpić, mając na uwadze nauczanie bioetyki katolickiej?
15. Decyzja o włączeniu wentylacji przewlekłej powinna być podjęta w specjalistycznym oddziale, po wnikliwej ocenie stanu zdrowia dziecka przez zespół, którego członkami, jeśli to jest możliwe, powinni być: pediatra, anestezjolog dziecięcy, neurolog dziecięcy, pulmonolog dziecięcy i gastroenterolog dziecięcy.
16. Postępując zgodnie ze standardami opieki nad dzieckiem z SMA, trzeba natychmiast podjąć wentylację, jeśli wystąpiła u niego niewydolność oddechowa. Należy jednak rozważyć, czy dziecku wystarczy jeszcze wentylacja „nieinwazyjna” (maska wentylacyjna nosowa), czy należy zastosować już wentylację „inwazyjną”. Niestety, z każdą z tych metod związane są również powikłania.
17. Według standardów, w opisanym przypadku niemowlę z SMA typu 1. w chwili wystąpienia niewydolności oddechowej oraz w jej następstwie niewydolności krążenia, powinno mieć włączoną sztuczną wentylację „inwazyjną” i wykonaną przezskórną endoskopową gastrostomię (PEG – Percutaneous Endoscopic Gastrostomy). Sztuczna wentylacja i leczenie żywieniowe, przy obecnych możliwościach medycyny, i w tym przypadku, są środkami proporcjonalnymi i podtrzymującymi życie, które jednak z dużym prawdopodobieństwem nie będzie trwało długo.
18. Odstąpienie od zabiegów nieproporcjonalnych, określanych jako terapia daremna, nie powinna budzić żadnych wątpliwości wśród członków zespołu medycznego.
19. W sytuacji, kiedy są wątpliwości, czy stosowane zabiegi są już daremną terapią, czy jeszcze nie, należy postępować na korzyść życia i zdrowia, kontynuując dotychczasową terapię i opiekę oraz wykorzystując dostępne i proporcjonalne środki, a do takich należy obecnie zaliczyć sztuczną wentylację i leczenie żywieniowe.
20. Należy jednak zastrzec, że w szczególnych przypadkach sztuczna wentylacja i leczenie żywieniowe, jak każdy inny środek medyczny lub technika, mogą być w swoich skutkach działaniem proporcjonalnym lub nieproporcjonalnym do sytuacji zdrowotnej pacjenta.
21. Miarą proporcjonalności danego środka, czy medycznej techniki jest nie tylko poprawa zdrowia, czy jakości życia, ale w przypadku opieki paliatywnej również umniejszanie cierpienia fizycznego i psychicznego.
22. Zastępczą wentylację należy stosować, jeśli jest nadzieja na uratowanie życia i poprawę jego jakości w przypadku opieki długoterminowej. Natomiast w przypadku opieki paliatywnej wówczas, jeśli sztuczna wentylacja zmniejszy, a przynajmniej nie zwiększy lęku i cierpienia spowodowanego dusznością.
23. Należy zrezygnować z wprowadzenia zastępczej wentylacji jako środka nieproporcjonalnego do obecnej sytuacji pacjenta, jeżeli do tej pory nie była wykorzystana, a zastosowanie jej wydłużyłoby trwające już umieranie (agonię) i nasiliło związane z nim cierpienie.
24. W tym przypadku celem rezygnacji ze sztucznej wentylacji nie jest przyspieszenie śmierci (eutanazja), ale powstrzymanie się od tej techniki wspomagającej życie, ponieważ rozpoczął się już nieuchronny proces umierania, a śmierć nastąpi w krótkim czasie i już nie można jej powstrzymać.
25. Podjęcie decyzji o rezygnacji wymaga dokonania istotnego rozróżnienia pomiędzy rozpoczętym już umieraniem, które upoważnia do odstąpienia od sztucznej wentylacji, a umieraniem dopiero przewidywanym w przyszłości, które nie daje podstaw do odstąpienia od sztucznej wentylacji.
26. Wycofanie się z już stosowanej wentylacji zastępczej jest moralnie usprawiedliwione w sytuacji stwierdzenia śmierci mózgu.
27. Oceny sytuacji, czy sztuczna wentylacja jest środkiem proporcjonalnym do stanu dziecka chorego na SMA, dokonuje zespół lekarzy. Kierownik (Ordynator) wraz ze współpracownikami powinien udźwignąć odpowiedzialność moralną za tę bardzo trudną decyzję. W szczególnie trudnych przypadkach jest wskazane skorzystanie z pomocy Szpitalnej Komisji ds. Daremnej Terapii.
28. Wola rodziców lub prawnych opiekunów, w podejmowaniu decyzji dotyczących sposobów leczenia dziecka z SMA, stanowi ważny, ale nie jedyny punkt odniesienia, pod warunkiem, że jest świadomie wyrażona i nie występuje przeciwko życiu i zdrowiu dziecka. Wymaga to wcześniejszego poinformowania ich, w sposób zrozumiały i wyczerpujący, o wszystkich możliwych metodach postępowania z dzieckiem, jak również przewidywanych skutkach korzystnych i niekorzystnych dla chorego.
29. Rozstrzyganie spraw spornych dotyczących terapii na drodze sądowej, jest ostatecznością, gdy zawiodą wszystkie inne środki. Dialog z rodzicami i troskliwa kompetentna opieka nad chorym dzieckiem są najlepszymi środkami do wypracowania właściwych decyzji.
30. Rodzina chorego dziecka powinna być otoczona wielospecjalistyczną opieką medyczną, psychologiczną, duszpasterską, prawną i socjalną w szpitalu i w domu – pozostając pod opieką zespołu domowej opieki hospicyjnej.
31. Pobyt w stacjonarnym ośrodku dla dzieci przewlekle wentylowanych jest ostatecznością, gdy rodzina nie potrafi, nie może lub odmawia opieki nad ciężko chorym dzieckiem.
32. Należy jednak wystrzegać się przerzucania na rodziców dziecka lub jego prawnych opiekunów całej odpowiedzialności za podjęcie decyzji o rezygnacji z daremnej terapii, gdyż takie postępowanie może być nieuprawnione, dla nich zbyt trudne do uniesienia i naznaczone daleko idącym subiektywizmem.
33. W przypadku stwierdzenia, że śmierć zbliża się nieuchronnie, a terapia wyczerpuje wszelkie znamiona daremności, tak naprawdę nie mamy już do czynienia z podejmowaniem decyzji, ale tylko z akceptacją ludzkiego losu. Wówczas pozostaje już tylko zapewnienie umierającemu dziecku godnych warunków umierania i łagodzenie cierpienia dostępnymi środkami oraz otoczenie jego rodziny opieką.
W imieniu Zespołu Ekspertów KEP ds. Bioetycznych
+ Józef Wróbel SCJ
Przewodniczący Zespołu
Warszawa, 19 marca 2019 roku